
Causes of Alkaptonuria
Early Causes of Alkaptonuria are commonly due to mutations in the HGD gene, located on chromosome 3. This gene provides instructions for producing the homogentisate 1,2-dioxygenase enzyme, which plays a critical role in breaking down the amino acids tyrosine and phenylalanine. When this enzyme is absent or malfunctioning, homogentisic acid (HGA) cannot be processed and starts to accumulate in the body.
Genetic Inheritance
Alkaptonuria follows an autosomal recessive inheritance pattern. This means:
- Both parents must carry one copy of the mutated gene.
- Each child has a 25% chance of inheriting the condition.
- Carriers usually remain symptom-free.
Because it is recessive, alkaptonuria often goes unnoticed in families until a child develops symptoms.
Homogentisic Acid and Ochronosis
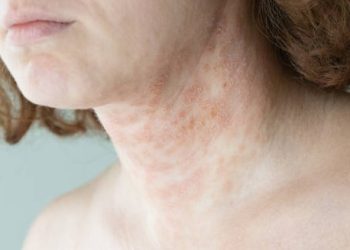
The excessive HGA that builds up over time is deposited in bones, cartilage, and connective tissues. Additionally, this pigment-like substance causes a bluish-black discolouration known as ochronosis, particularly visible in ear cartilage, eye whites (sclera), and skin folds.
Over decades, this deposition leads to joint stiffness, spinal degeneration, and early-onset osteoarthritis.
Global and Local Prevalence
Alkaptonuria is extremely rare, affecting roughly 1 in 250,000 to 1 in 1 million people worldwide. Furthermore, while cases in South Africa are rare, underdiagnosis is possible due to a lack of awareness.
Research suggests it may be more common in certain populations, such as in Slovakia or parts of the Dominican Republic, where gene mutations have been passed through generations.
Although it’s a lifelong condition, understanding its genetic basis allows for earlier recognition. Furthermore, genetic counselling, and planning supportive care strategies.
👉 [Next: Diagnosis of Alkaptonuria]