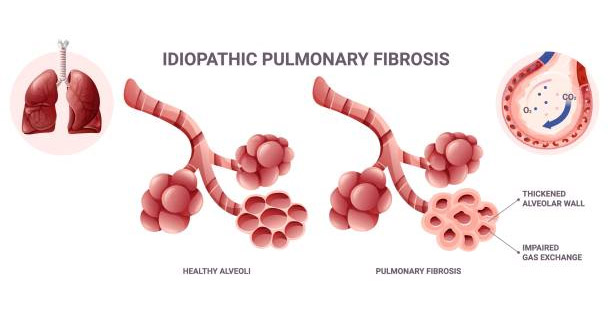

Diagnosis of idiopathic pulmonary fibrosis is often complex and requires a multifaceted approach to rule out other conditions that may mimic its symptoms.
Why Diagnosis Is Difficult
Idiopathic pulmonary fibrosis (IPF) looks similar to many other lung diseases, especially interstitial lung disease (ILD). This makes accurate diagnosis challenging. Early symptoms such as breathlessness and tiredness are often mild. Many people ignore them or think they are due to aging. Still, an early and correct diagnosis of idiopathic pulmonary fibrosis is critical for starting treatment and slowing the disease.
Medical History and Physical Exam
The first step in diagnosing IPF starts with a detailed medical history and physical check-up. Doctors ask about long-term symptoms like dry cough and shortness of breath. They also ask about exposure to dust, smoke, or chemicals, past smoking habits, family history, and any autoimmune diseases. These details help rule out other lung problems that may need different treatment.
During the exam, a doctor may hear “Velcro-like” crackles in the lungs with a stethoscope. These soft sounds happen when a person breathes in and often suggest IPF. Another sign is finger clubbing, where fingertips become round and swollen because of low oxygen. These clues do not confirm IPF, but they make doctors more suspicious when combined with symptoms and history.
Imaging Tests
To confirm IPF, doctors usually order a high-resolution CT scan (HRCT). This special scan shows detailed images of lung tissue. In IPF, HRCT often shows a usual interstitial pneumonia (UIP) pattern. This includes honeycombing, thickened tissue, and airway changes. When this pattern is clear, doctors can often confirm IPF without other invasive tests.
When Imaging Is Not Enough
If the scan does not clearly show UIP, doctors may suggest a lung biopsy. This can be done by surgery or with a less invasive cryobiopsy. A small piece of lung tissue is studied under a microscope to confirm IPF. But biopsies carry risks, especially in older patients or those with poor lung function. Doctors weigh these risks before deciding.
Pulmonary Function Tests
Another key step is lung function testing. These tests check how well the lungs move air and transfer oxygen. IPF patients often have stiff lungs that cannot expand well. This shows as low forced vital capacity (FVC) and poor oxygen transfer (DLCO). Repeat tests over time help track disease progress.
Blood Tests and Other Checks
Blood tests rule out other diseases that look like IPF, such as autoimmune disorders. They check for antibodies and inflammation markers. But no blood test can confirm IPF yet. Researchers are studying new markers, but these are not in routine use now.
Role of Multidisciplinary Teams
Because IPF is hard to diagnose, many patients are referred to a team of specialists. This team often includes lung doctors, radiologists, pathologists, and sometimes rheumatologists. They review scans, lab tests, and symptoms together. This combined approach is now the best way to confirm IPF and plan the right treatment.
Bronchoalveolar Lavage (BAL)
Sometimes doctors use BAL to collect lung fluid for testing. It cannot confirm IPF on its own, but it helps rule out infections or other lung diseases. It is most useful in younger patients or when the case is unclear.
Importance of Early Diagnosis
Getting diagnosed early makes a big difference in treatment and quality of life. Sadly, many patients face long delays. Symptoms are often blamed on age or asthma. Lack of specialist access also slows diagnosis. Raising awareness among doctors and patients can help reduce these delays.
Final Thoughts on Diagnosis of Idiopathic Pulmonary Fibrosis
The diagnosis of idiopathic pulmonary fibrosis needs a full review of symptoms, scans, lung tests, and sometimes biopsies. A specialist team often gives the most accurate answer. The sooner IPF is found, the sooner treatment can begin to manage symptoms and improve life quality.